Quantifying transmission intensity and heterogeneity is crucial to ascertain the threat posed by infectious diseases and inform the design of interventions. Methods that jointly estimate the reproduction number R and the dispersion parameter k have however mainly remained limited to the analysis of epidemiological clusters or contact tracing data, whose collection often proves difficult. Here, we show that clusters of identical sequences are imprinted by the pathogen offspring distribution, and we derive an analytical formula for the distribution of the size of these clusters. We develop and evaluate a novel inference framework to jointly estimate the reproduction number and the dispersion parameter from the size distribution of clusters of identical sequences. We then illustrate its application across a range of epidemiological situations. Finally, we develop a hypothesis testing framework relying on clusters of identical sequences to determine whether a given pathogen genetic subpopulation is associated with increased or reduced transmissibility. Our work provides new tools to estimate the reproduction number and transmission heterogeneity from pathogen sequences without building a phylogenetic tree, thus making it easily scalable to large pathogen genome datasets.
Quantifying transmission intensity and heterogeneity is crucial to ascertain the threat posed by infectious diseases and inform the design of interventions. Methods that jointly estimate the reproduction number R and the dispersion parameter k have however mainly remained limited to the analysis of epidemiological clusters or contact tracing data, whose collection often proves difficult. Here, we show that clusters of identical sequences are imprinted by the pathogen offspring distribution, and we derive an analytical formula for the distribution of the size of these clusters. We develop and evaluate a novel inference framework to jointly estimate the reproduction number and the dispersion parameter from the size distribution of clusters of identical sequences. We then illustrate its application across a range of epidemiological situations. Finally, we develop a hypothesis testing framework relying on clusters of identical sequences to determine whether a given pathogen genetic subpopulation is associated with increased or reduced transmissibility. Our work provides new tools to estimate the reproduction number and transmission heterogeneity from pathogen sequences without building a phylogenetic tree, thus making it easily scalable to large pathogen genome datasets.
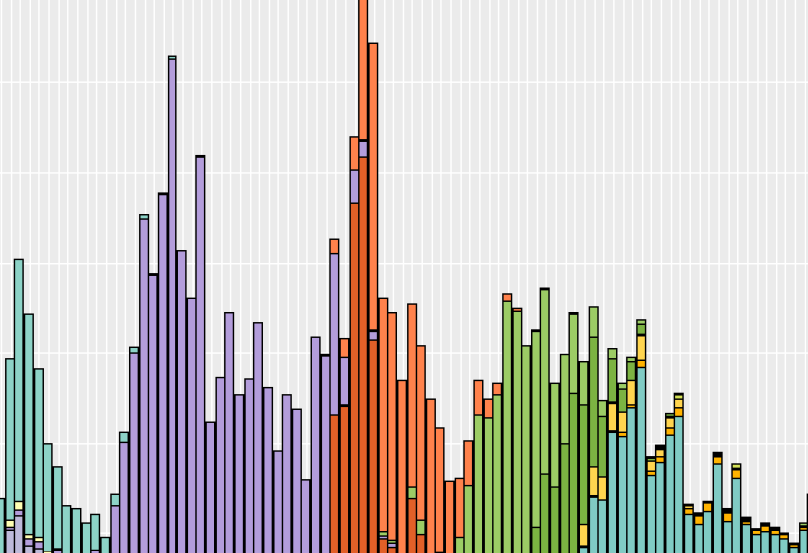
Washington Department of Health's Sequencing and Variants Report for the month of May, 2023.
Just trying out some of the formatting options as well.
X2t
Can I create markdown `code` here?
<beast\>for thing in things:
do x
do y
endMore text
More text
| Row 1 | thing | thing | thing |
| Row 2 | thing | thing | thing |